New computational tool may speed drug discovery
November 2, 2016
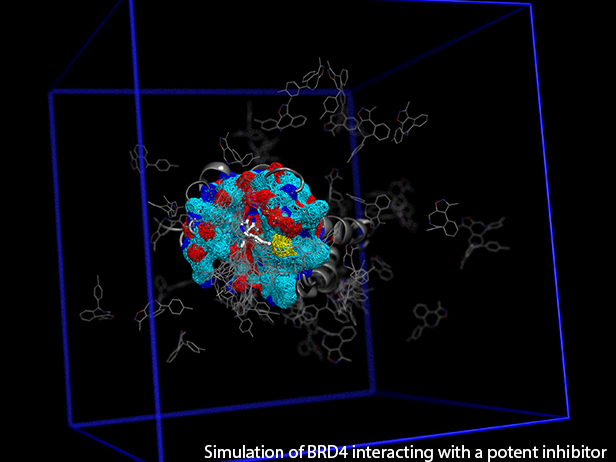
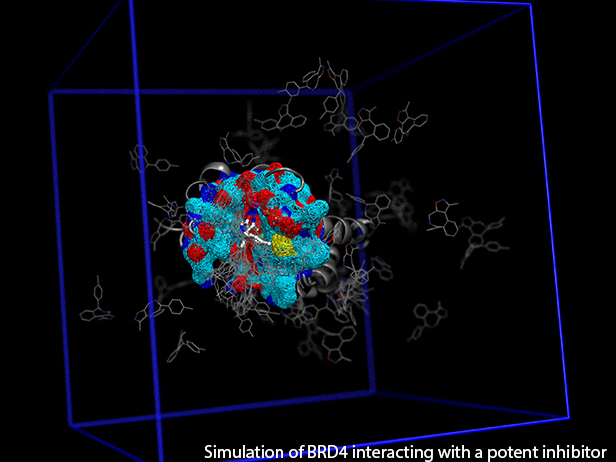
GRAND RAPIDS, Mich. (November 3, 2016)—A new computational tool called fABMACS is helping scientists see beyond static images of proteins to more efficiently understand how these molecules function, which could ultimately speed up the drug discovery process.
Proteins are the molecular workhorses of biology—they carry out the instructions written in the genetic code. Their shape plays a crucial role in their function and their ability to interact with other molecules. Scientists study these interactions to develop new insights into protein function and to develop targeted therapies for diseases such as cancer.
“The goal of targeted drug design is to create a molecule that interacts specifically with a protein, and this requires a description of protein-drug interactions that is precise—down to the placement of each atom,” said Bradley Dickson, Ph.D., a computational biophysicist in the laboratory of Scott Rothbart, Ph.D., at Van Andel Research Institute (VARI) and first author of a paper describing the tool. “The creation of fABMACS is a significant step toward robust virtual drug discovery because it saves time and money. It allows us to better harness the power of existing software while greatly improving our ability to predict the way that a potential drug interacts with a protein.”
Scientists often rely on collecting snapshots of proteins to determine how they may interact with a potential drug. However, these images are static and do not depict changes in proteins’ shape.
“These snapshots provide valuable insight that can be enriched by fABMACS,” said Rothbart, assistant professor at VARI and the study’s senior author. “fABMACS allows us to simulate chemical changes to the drug and more quickly predict how those changes impact its interaction with the target protein. Ultimately, this could translate to improved drug potency and efficacy.”
To demonstrate the tool’s capabilities, the team ran several accelerated computer simulations of the epigenetic regulatory protein BRD4 bound to a drug that is currently in phase I clinical trials for blood cancers. They demonstrated that a slight change to the compound’s chemical structure could improve binding to its target protein, thereby improving its effect. The results of this work were published recently in the Journal of Chemical Physics.
Fast, stable and scalable
fABMACS is an add-on to existing molecular dynamics software. It is based on GROMACSv5.0.5 and optimizes network communication and load balancing—both critical aspects of software development in parallel computing environments—to achieve a low-overhead implementation of new free-energy techniques. fABMACS also comes with a built-in configuration tool that allows the code to be tailored to different applications without requiring the user to manually edit the code, which maximizes transferability.
fABMACS is free to download under a GNU General Public License. More information may be found at rothbartlab.vai.org/tools.
Research reported in this publication was supported by VARI and by the National Cancer Institute of the National Institutes of Health under Award Number R00CA181343. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
###
Dickson BM, de Waal P, Ramjan Z, Xu HE, Rothbart SB. 2016. A fast, open source implementation of adaptive biasing potentials uncovers a ligand design strategy for the chromatin regulator BRD4. J Chem Phys.
Media Contact
Beth Hinshaw Hall, [email protected], 616.234.5519