
News
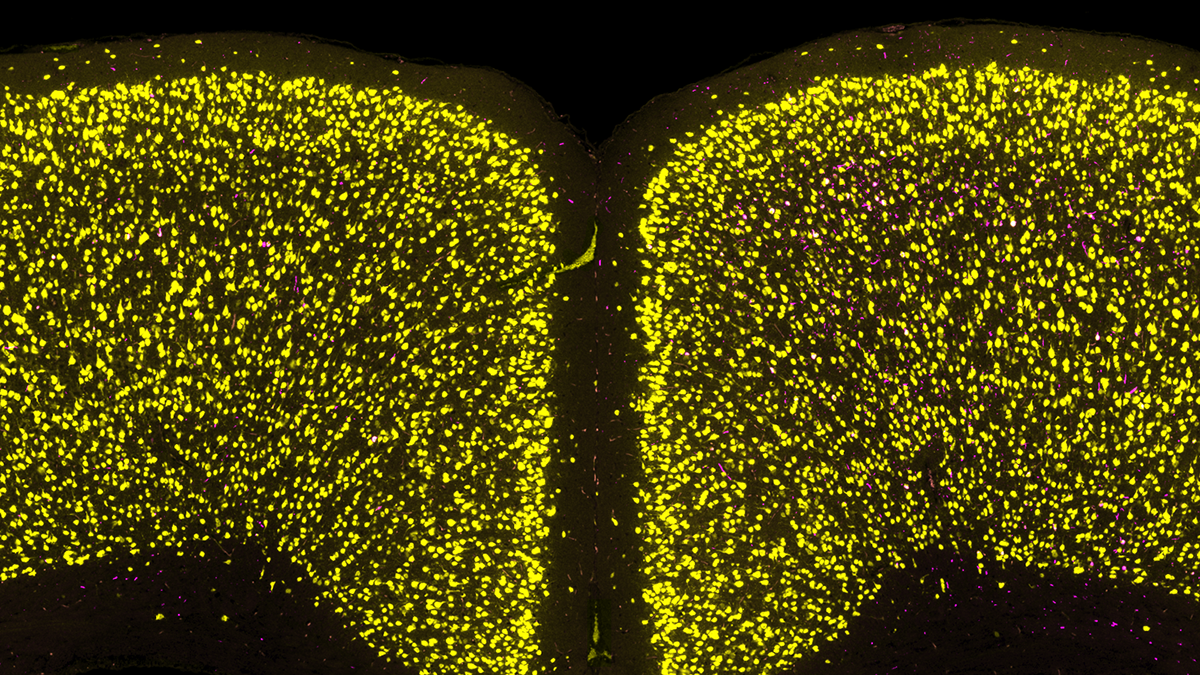
Scientists identify cell vulnerability ‘fingerprint’ related to Parkinson’s, Lewy body dementia

Van Andel Institute to recognize eminent Parkinson’s disease scientist Dr. Anders Björklund with 2024 Jay Van Andel Award
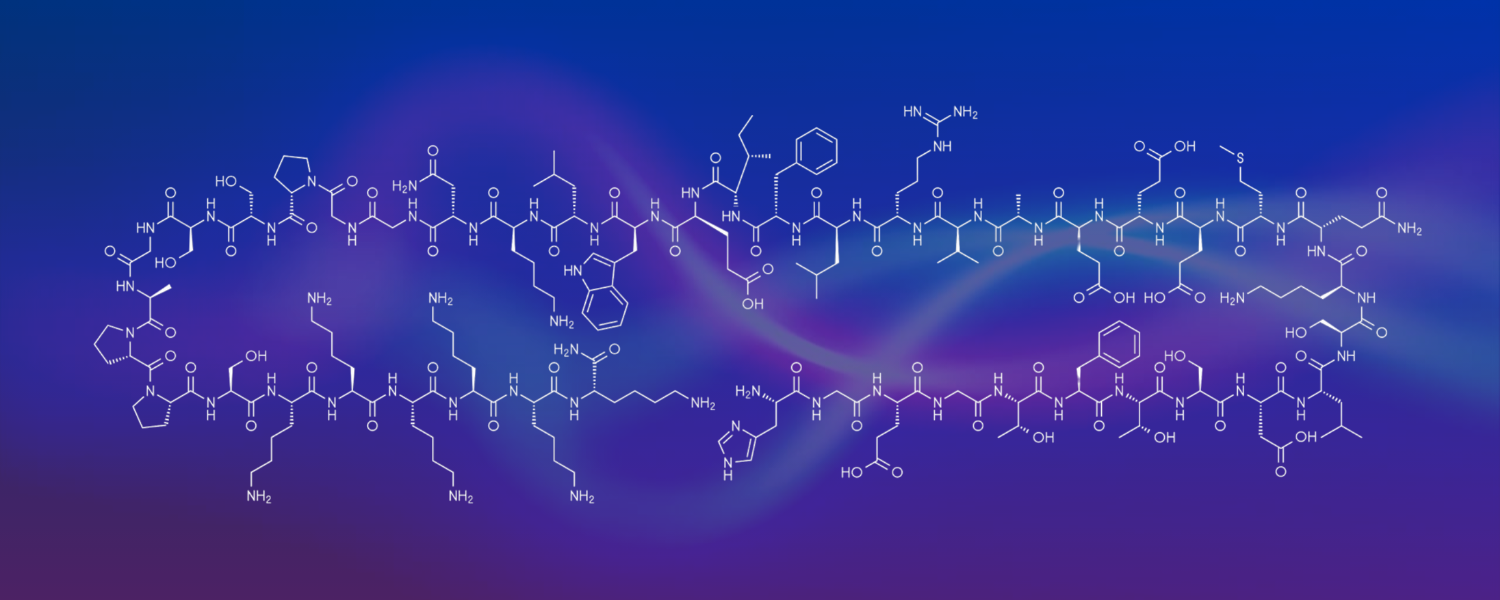
Phase 2 clinical trial of Type 2 diabetes drug for treatment of Parkinson’s shows positive and promising results

Exploring the relationship between nutrition and cancer
Exploring the relationship between nutrition and cancer
Tuberculosis is an ancient problem. Modern technology is helping solve it

Supporting a shared mission

How a ‘blockbuster’ treatment for diabetes and obesity might slow or stop Parkinson’s
Scientists identify cell vulnerability ‘fingerprint’ related to Parkinson’s, Lewy body dementia
Van Andel Institute to recognize eminent Parkinson’s disease scientist Dr. Anders Björklund with 2024 Jay Van Andel Award
Phase 2 clinical trial of Type 2 diabetes drug for treatment of Parkinson’s shows positive and promising results
Combining epigenetic cancer medications may have benefit for colorectal cancers and other tumor types
We’re here to help you tell your story.
Van Andel Institute is home to experts in a range of fields, including epigenetics, neurodegeneration, metabolism, structural and cellular biology, and K–12 STEM education.
The Institute’s Communications and Marketing Department assists journalists with information about the groundbreaking research happening at VAI. We can arrange interviews with VAI scientists and provide supporting materials, such as fact sheets, photographs and b-roll video to support the production of news packages. For press inquiries, please reach out to us at [email protected] or submit an inquiry below.
Media contacts
Beth Hinshaw, M.S.
Director, Communications & Marketing
616.234.5519
[email protected]
Zane McMillin
Content Strategist, Communications & Marketing
[email protected]